Hello,
I tried to connect ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/ , but I got a "530 Login incorrect".
Would you please check that?
Thanks for your help,
Henry
Hello,
I tried to connect ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/ , but I got a "530 Login incorrect".
Would you please check that?
Thanks for your help,
Henry
Test that Queue is correctly installed, and that the supporting tools like Java are in your path.
The command we're going to run is a very simple command that asks Queue to print out a list of available command-line arguments and options. It is so simple that it will ALWAYS work if your Queue package is installed correctly.
Note that this command is also helpful when you're trying to remember something like the right spelling or short name for an argument and for whatever reason you don't have access to the web-based documentation.
Type the following command:
java -jar <path to Queue.jar> --help
replacing the <path to Queue.jar> bit with the path you have set up in your command-line environment.
You should see usage output similar to the following:
usage: java -jar Queue.jar -S <script> [-jobPrefix <job_name_prefix>] [-jobQueue <job_queue>] [-jobProject <job_project>]
[-jobSGDir <job_scatter_gather_directory>] [-memLimit <default_memory_limit>] [-runDir <run_directory>] [-tempDir
<temp_directory>] [-emailHost <emailSmtpHost>] [-emailPort <emailSmtpPort>] [-emailTLS] [-emailSSL] [-emailUser
<emailUsername>] [-emailPass <emailPassword>] [-emailPassFile <emailPasswordFile>] [-bsub] [-run] [-dot <dot_graph>]
[-expandedDot <expanded_dot_graph>] [-startFromScratch] [-status] [-statusFrom <status_email_from>] [-statusTo
<status_email_to>] [-keepIntermediates] [-retry <retry_failed>] [-l <logging_level>] [-log <log_to_file>] [-quiet]
[-debug] [-h]
-S,--script <script> QScript scala file
-jobPrefix,--job_name_prefix <job_name_prefix> Default name prefix for compute farm jobs.
-jobQueue,--job_queue <job_queue> Default queue for compute farm jobs.
-jobProject,--job_project <job_project> Default project for compute farm jobs.
-jobSGDir,--job_scatter_gather_directory <job_scatter_gather_directory> Default directory to place scatter gather
output for compute farm jobs.
-memLimit,--default_memory_limit <default_memory_limit> Default memory limit for jobs, in gigabytes.
-runDir,--run_directory <run_directory> Root directory to run functions from.
-tempDir,--temp_directory <temp_directory> Temp directory to pass to functions.
-emailHost,--emailSmtpHost <emailSmtpHost> Email SMTP host. Defaults to localhost.
-emailPort,--emailSmtpPort <emailSmtpPort> Email SMTP port. Defaults to 465 for ssl,
otherwise 25.
-emailTLS,--emailUseTLS Email should use TLS. Defaults to false.
-emailSSL,--emailUseSSL Email should use SSL. Defaults to false.
-emailUser,--emailUsername <emailUsername> Email SMTP username. Defaults to none.
-emailPass,--emailPassword <emailPassword> Email SMTP password. Defaults to none. Not
secure! See emailPassFile.
-emailPassFile,--emailPasswordFile <emailPasswordFile> Email SMTP password file. Defaults to none.
-bsub,--bsub_all_jobs Use bsub to submit jobs
-run,--run_scripts Run QScripts. Without this flag set only
performs a dry run.
-dot,--dot_graph <dot_graph> Outputs the queue graph to a .dot file. See:
http://en.wikipedia.org/wiki/DOT_language
-expandedDot,--expanded_dot_graph <expanded_dot_graph> Outputs the queue graph of scatter gather to
a .dot file. Otherwise overwrites the
dot_graph
-startFromScratch,--start_from_scratch Runs all command line functions even if the
outputs were previously output successfully.
-status,--status Get status of jobs for the qscript
-statusFrom,--status_email_from <status_email_from> Email address to send emails from upon
completion or on error.
-statusTo,--status_email_to <status_email_to> Email address to send emails to upon
completion or on error.
-keepIntermediates,--keep_intermediate_outputs After a successful run keep the outputs of
any Function marked as intermediate.
-retry,--retry_failed <retry_failed> Retry the specified number of times after a
command fails. Defaults to no retries.
-l,--logging_level <logging_level> Set the minimum level of logging, i.e.
setting INFO get's you INFO up to FATAL,
setting ERROR gets you ERROR and FATAL level
logging.
-log,--log_to_file <log_to_file> Set the logging location
-quiet,--quiet_output_mode Set the logging to quiet mode, no output to
stdout
-debug,--debug_mode Set the logging file string to include a lot
of debugging information (SLOW!)
-h,--help Generate this help message
If you see this message, your Queue installation is ok. You're good to go! If you don't see this message, and instead get an error message, proceed to the next section on troubleshooting.
Let's try to figure out what's not working.
First, make sure that your Java version is at least 1.6, by typing the following command:
java -version
You should see something similar to the following text:
java version "1.6.0_12"
Java(TM) SE Runtime Environment (build 1.6.0_12-b04)
Java HotSpot(TM) 64-Bit Server VM (build 11.2-b01, mixed mode)
If the version is less then 1.6, install the newest version of Java onto the system. If you instead see something like
java: Command not found
make sure that java is installed on your machine, and that your PATH variable contains the path to the java executables.
On a Mac running OS X 10.5+, you may need to run /Applications/Utilities/Java Preferences.app and drag Java SE 6 to the top to make your machine run version 1.6, even if it has been installed.
I am processing single-cell RNAseq data which I downloaded using GEO accession number (it was in .sra format which I converted to .bam)
Now I'm trying to run the scRNAseq pipeline and got stuck since it seems like I don't have read groups in the header.
I'm trying to use Picard's AddOrReplaceReadGroups with the following command:
java -Xmx15g picard.jar AddOrReplaceReadGroups \
I=SRR5164436.bam \
O=SRR5164436_RG.bam \
RGID=bam1 \
RGLB=lib1 \
RGPL=illumina \
RGPU=ad_lib_Chow1 \
RGSM=sra36
But I get this error:
Exception in thread "main" htsjdk.samtools.SAMFormatException: SAM validation error: ERROR: Record 1, Read name 1, RG ID on SAMRecord not found in header: 1
I don't understand why it is happening. Can you please help? See below the complete error message.
Also, can you please explain where can I get the RGPU, if I don't have the .fastq file? If I cannot, I'd have to put some arbitrary number?
11:51:55.904 INFO NativeLibraryLoader - Loading libgkl_compression.so from jar:file:/cvar/jhlab/Kathy/Drop-seq/picard-2.12.2/picard.jar!/com/intel/gkl/native/libgkl_compression.so
[Mon Oct 23 11:51:55 EDT 2017] AddOrReplaceReadGroups INPUT=SRR5164436.bam OUTPUT=SRR5164436_RG.bam RGID=bam1 RGLB=lib1 RGPL=illumina RGPU=ad_lib_Chow1 RGSM=sra36 VERBOSITY=INFO QUIET=false VALIDATION_STRINGENCY=STRICT COMPRESSION_LEVEL=5 MAX_RECORDS_IN_RAM=500000 CREATE_INDEX=false CREATE_MD5_FILE=false GA4GH_CLIENT_SECRETS=client_secrets.json USE_JDK_DEFLATER=false USE_JDK_INFLATER=false
[Mon Oct 23 11:51:55 EDT 2017] Executing as kushakov@uger-c065.broadinstitute.org on Linux 2.6.32-696.6.3.el6.x86_64 amd64; Java HotSpot(TM) 64-Bit Server VM 1.8.0_121-b13; Deflater: Intel; Inflater: Intel; Picard version: 2.12.2-SNAPSHOT
INFO 2017-10-23 11:51:55 AddOrReplaceReadGroups Created read group ID=bam1 PL=illumina LB=lib1 SM=sra36
[Mon Oct 23 11:51:56 EDT 2017] picard.sam.AddOrReplaceReadGroups done. Elapsed time: 0.00 minutes.
Runtime.totalMemory()=2058354688
To get help, see http://broadinstitute.github.io/picard/index.html#GettingHelp
Exception in thread "main" htsjdk.samtools.SAMFormatException: SAM validation error: ERROR: Record 1, Read name 1, RG ID on SAMRecord not found in header: 1
at htsjdk.samtools.SAMUtils.processValidationErrors(SAMUtils.java:454)
at htsjdk.samtools.BAMFileReader$BAMFileIterator.advance(BAMFileReader.java:812)
at htsjdk.samtools.BAMFileReader$BAMFileIterator.(BAMFileReader.java:783)
at htsjdk.samtools.BAMFileReader$BAMFileIterator.(BAMFileReader.java:771)
at htsjdk.samtools.BAMFileReader.getIterator(BAMFileReader.java:474)
at htsjdk.samtools.SamReader$PrimitiveSamReaderToSamReaderAdapter.iterator(SamReader.java:478)
at picard.sam.AddOrReplaceReadGroups.doWork(AddOrReplaceReadGroups.java:141)
at picard.cmdline.CommandLineProgram.instanceMain(CommandLineProgram.java:268)
at picard.cmdline.PicardCommandLine.instanceMain(PicardCommandLine.java:98)
at picard.cmdline.PicardCommandLine.main(PicardCommandLine.java:108)
Dear gatk team,
I am struggle with an issue, it's 4 day that I try to access the resource bundle without success.
I am following the steps posted in the page, as username: gsapubftp-anonymous and as password blank space, the window thats open when I try to log in tremble as if the password was incorrect.
I also try from command line with wget but the connection get interrupted.
I use a Yosemite OS X but I also try with El Capitan and a windows 10 PC, also from different networks.
Any suggestion?
Best
Luca
In fact, going forward all of our new projects will use GRCh38. During this transition over the coming year, we will keep supporting GRCh37/hg19. Here are nine takeaways to help you get started in using the latest reference.
 Don’t know alternate contig from alternate dimension? Spend five minutes now to review terminology in our Dictionary entry Reference Genome Components. At the least, you should understand the distinction between the primary assembly and alternate contigs.
Don’t know alternate contig from alternate dimension? Spend five minutes now to review terminology in our Dictionary entry Reference Genome Components. At the least, you should understand the distinction between the primary assembly and alternate contigs.
Long BAM headers notwithstanding, GRCh38 alternate contig sequences are only ~3.6% of the primary assembly length (see table). They encompass alternate haplotypes for which we cannot easily represent variants on the primary assembly. According to my estimation, roughly a tenth of a percent (101,845 basepairs) of the alternate sequence appears highly divergent.
Download your own analysis reference set from the GATK resource bundle. Be certain you are mapping to a version of the genome that hard-masks--replaces with Ns--Y chromosome PARs. Imagine the SHOX of not being able to call variants for pseudoautosomal regions.
Conceptually it rewraps and regifts the challenge of calling variants for paralogous regions of the genome. The difference is that alternate contigs encompass sequence that is homologous as well as highly divergent for loci across a population instead of across a genome. By definition, we cannot easily represent the variants alternate haplotypes generate against the primary assembly. And so GRCh38 arms us with named alternate contigs that beg to be used when we call their variants. How folks choose to do this with the leeway given by VCF specifications will depend on research aims.
 You want to map in an alt-aware manner, i.e. you want your alts handled. Without the handling, you’ll just get a bunch of MAPQ zero ghost reads mapping to both (i) the primary assembly regions that have alternate contigs and (ii) the homologous alternate contig regions. Just as you cannot eat ghost chips, GATK tools refuse to consider zero (and low) MAPQ alignments. No. You. Do. Not. Want. This. Make sure to update to BWA-MEM version 0.7.13+ to be able to map with alt-handling. I’m partial to calling it ghost-busting. This enables two things. First, because it prioritizes alignments on the primary assembly by disappearing alignments from the alternate contigs, it effectively lets you avoid redundantly calling variants on homologous regions of alternate loci. Second, it allows for an additional postalt-processing step that populates multiple alt loci contig(s) with nonzero MAPQ alignments. This enables super-charged variant calling on all the alt contigs. For details, read BWA’s alt-specific README-alt. Although the README currently is marked for an earlier version of the tool, its concepts still apply.
You want to map in an alt-aware manner, i.e. you want your alts handled. Without the handling, you’ll just get a bunch of MAPQ zero ghost reads mapping to both (i) the primary assembly regions that have alternate contigs and (ii) the homologous alternate contig regions. Just as you cannot eat ghost chips, GATK tools refuse to consider zero (and low) MAPQ alignments. No. You. Do. Not. Want. This. Make sure to update to BWA-MEM version 0.7.13+ to be able to map with alt-handling. I’m partial to calling it ghost-busting. This enables two things. First, because it prioritizes alignments on the primary assembly by disappearing alignments from the alternate contigs, it effectively lets you avoid redundantly calling variants on homologous regions of alternate loci. Second, it allows for an additional postalt-processing step that populates multiple alt loci contig(s) with nonzero MAPQ alignments. This enables super-charged variant calling on all the alt contigs. For details, read BWA’s alt-specific README-alt. Although the README currently is marked for an earlier version of the tool, its concepts still apply.
Special handling requires a special index file. Alt-handling requires that an ALT index is available with the other BWA indexes. Heng Li provides the ALT index for GRCh38 in the linux bwa.kit v0.7.15. Find the hs38DH.fa.alt file in the resource-GRCh38 folder and explore it using Samtools to confirm the following.
Each alternate contig record lists a CIGAR string, some of which are rather convoluted, that aligns the alternate contig back to its primary assembly locus. For six of the alternate contigs, we have two alignments each.
The decoys contain transposable and alpha satellite elements including diverged variants. Why are they represented in the ALT index? See the next takeaway.
Tutorial#8017 starts with indexing the reference, reiterates the essentiality of the ALT index and then maps in an alt-aware manner using simulated reads to a miniature-reference. It then goes on to show how to postalt-process alignments using the bwa-postalt.js script. The tutorial does not tell you what to do per se, but rather shows what happens when you use certain options. You definitely want to read sections 5–6 if you plan on calling variants on alternate contigs.
During postalt-processing, two reshufflings take place. First, alignments that can map to both a primary locus and an alternate locus are mapped to both with non-zero MAPQ alignments. These multimappers are supplementary on the alt. Second, if an alignment on the primary assembly aligns better on a decoy contig, then its alignment on the primary assembly is deprioritized with a zero MAPQ score. The tutorial gives an example of the first reshuffle. For those interested in seeing the second reshuffle, I have a suggestion. Change the mini-reference’s single ALT index record to mimic that of a decoy, i.e. change it to an unmapped record, then see what happens when you postalt-process.
If your research aims require one of the reshufflings but not the other, or selective handling for particular loci, then one approach could be to modify the ALT index for the selective postalt-processing.
Tutorial#7859 shows how to generate simulated reads so you can see results akin to those in Tutorial#8017 for your favorite alternate contig. For both tutorials, I use the GPI gene’s singular alternate contig as the example.
Using the liberty the blog format provides, I will digress here. The GPI locus encodes for glucose-6-phosphate isomerase, a protein that has an intracellular role in sugar metabolism and also moonlights extracellularly as Neuroleukin, a factor involved in nerve tissue growth. I chose this locus because (i) it is one of the smallest alternate contigs not near a telomere, (ii) I used to study metabolism and (iii) I worked on an identically named, unrelated molecule. Yes, really.
So, how significant are the alternate contigs? To start answering this question, I asked another. What story can I find for the GPI locus?
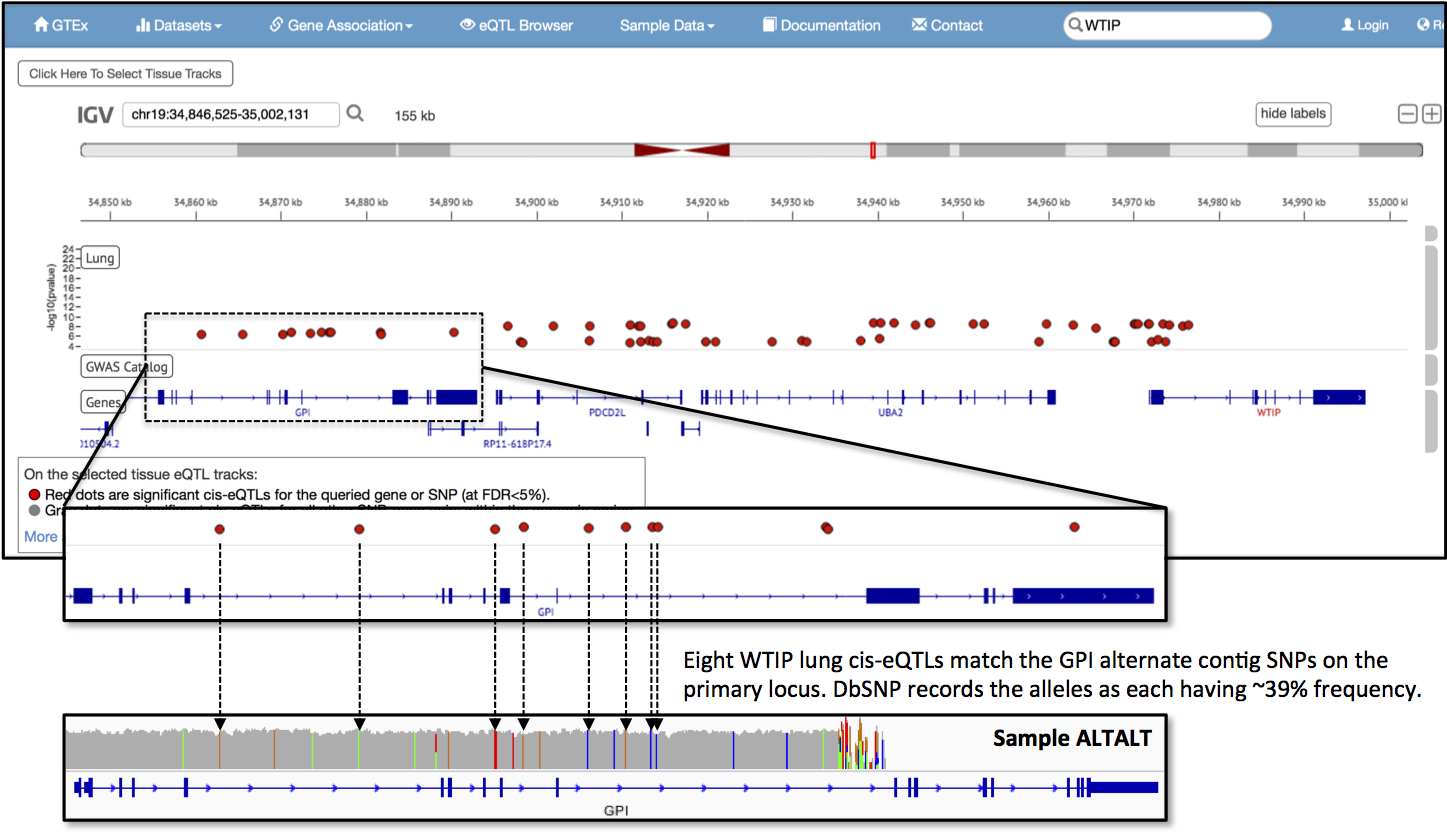 I did a little digging last Saturday afternoon for evidence of the alternate haplotype in data resources. In GTex, a project that measures healthy tissue-specific RNA isoform expression, I found that the GPI locus provides cis-eQTLs for WTIP in lung tissue. WTIP encodes for Wilms tumor 1 interacting protein and is three genes down from the GPI locus. Eight of the 11 eQTL sites on the GPI gene match SNPs that my simulated reads, representing the alternate haplotype, generate on the primary assembly. These sites, when I look them up in dbSNP, are all listed as minor alleles and intronic variants. The average global minor allele frequency for the eight SNPs is 38.7% (+/- 0.90%), with 1936 (+/- 45.0) observations in the 1000 Genomes Project phase 3 data. It looks like the GPI locus alternate haplotype is not uncommon and it already has some observed associations.
I did a little digging last Saturday afternoon for evidence of the alternate haplotype in data resources. In GTex, a project that measures healthy tissue-specific RNA isoform expression, I found that the GPI locus provides cis-eQTLs for WTIP in lung tissue. WTIP encodes for Wilms tumor 1 interacting protein and is three genes down from the GPI locus. Eight of the 11 eQTL sites on the GPI gene match SNPs that my simulated reads, representing the alternate haplotype, generate on the primary assembly. These sites, when I look them up in dbSNP, are all listed as minor alleles and intronic variants. The average global minor allele frequency for the eight SNPs is 38.7% (+/- 0.90%), with 1936 (+/- 45.0) observations in the 1000 Genomes Project phase 3 data. It looks like the GPI locus alternate haplotype is not uncommon and it already has some observed associations.
Check it out in our Broad pipelines WDL scripts repository. The document describing the workflow has the .md extension in the set named PairedEndSingleSampleWf. Even if you are unfamiliar with what is a WDL, no worries. The document focuses on explaining the data transformation steps from alignment to single-sample SNP and indel variant calling. The workflow maps paired reads in an alt-aware manner to GRCh38 and then uses HaplotypeCaller to generate a GVCF callset for the primary assembly. New features the workflow uses include query-grouped alignments through duplicate marking and addition of NM and UQ tags with SetNmAndUqTags.
It’s pretty straightforward. Using instructions provided by our WDL documentation, even yours truly has written her first three scripts for Tutorial#8017’s workflows. These we share via our new GATK Tutorials WDL scripts repo. WDL scripts will become more prevalent going forward. In conjunction with Docker, these process-centric pipeline scripts enable better provenance and reproducibility in research. If you are a complete newb to WDL, e.g. don’t know how to pronounce the acronym, then start with Blog#7349.
Want to help build our GRCh38 resources? Share your findings by posting a comment.
I just upgraded from GATK 3.7 to the newly released GATK 3.8 (3.8-0-ge9d806836) and I am getting a StatusLogger error:
ERROR StatusLogger Unable to create class org.apache.logging.log4j.core.impl.Log4jContextFactory specified in jar:file:/path/GenomeAnalysisTK-3.8-0/GenomeAnalysisTK.jar!/META-INF/log4j-provider.properties
ERROR StatusLogger Log4j2 could not find a logging implementation. Please add log4j-core to the classpath. Using SimpleLogger to log to the console...
Despite the error message, the tools seem to work just fine as far as I can tell.
Is this really an error? Is there a way to fix it?
Hi,
I tried to use GATK4 Mutect2 to call somatic mutations and found some weird MNP results:
CHROM POS REF ALT
chr2 157214886 GT TT
chr4 130772884 CGTGT TGTGT
chr4 145617203 TAAA AAAA
chr5 7857891 CAA AAA
chr5 30904821 AT TT
It sounds these variants should be simple SNPs however called as MNP here. Is it a bug in variant calling of Mutect2? Does GATK4 Mutect2 support MNP calling now?
Thanks!
Hi- I'm using Mutect2 from GenomeAnalysisTK-3.8-0-ge9d806836, everything looks good but I noticed that the vcf header of the output contains the INFO:
##INFO=<ID=PON,Number=1,Type=String,Description="Count from Panel of Normals">
I guess this field tells how many samples in the PON contain the variant, right? However, I never see the PON field ever used in the VCF records even if some variants are marked with the "panel_of_normals" filter, for example:
chr1 186341 . T G . panel_of_normals;t_lod_fstar ECNT=1;HCNT=10;MAX_ED=.;MIN_ED=.;NLOD=14.65;TLOD=5.22 GT:AD:AF:ALT_F1R2:ALT_F2R1:FOXOG:QSS:REF_F1R2:REF_F2R1 0/1:50,3:0.057:2:1:0.667:1542,92:22:28 0/0:66,1:0.015:0:1:0.00:2068,29:41:25
Is this expected?
And possibly related... The description of the "panel_of_normals" filter says: Seen in at least 2 samples in the panel of normals. However, I prepared the PON using --minN 1 which should activate the panel_of_normals filter for just one normal found. Is this just an oversight in the description or am I missing something?
Here's the synopsis of the relevant commands I used:
java -Xmx5g -jar ~/applications/gatk/GenomeAnalysisTK-3.8-0-ge9d806836/GenomeAnalysisTK.jar \
-T MuTect2 \
-R {params.ref} \
-I:tumor {input.tumour} \
-I:normal {input.normal} \
--normal_panel {input.pon} \
--dbsnp {params.dbsnp} \
--cosmic {params.cosmic} \
-L {params.chrom} \
--min_base_quality_score 20 \
--disable_auto_index_creation_and_locking_when_reading_rods \
-o {output.vcf}
and for PON:
java -Xmx8g -jar ~/applications/gatk/GenomeAnalysisTK-3.8-0-ge9d806836/GenomeAnalysisTK.jar \
-T CombineVariants \
-R {params.ref} \
{params.variant_str} \
-minN 1 \
--setKey "null" \
--filteredAreUncalled \
--filteredrecordsmergetype KEEP_IF_ANY_UNFILTERED \
-o pon/panelOfNormals.tmp.vcf
Thank you!
We recommend performing variant discovery in a way that enables joint analysis of multiple samples, as laid out in our Best Practices workflow. That workflow includes a joint analysis step that empowers variant discovery by providing the ability to leverage population-wide information from a cohort of multiple sample, allowing us to detect variants with great sensitivity and genotype samples as accurately as possible. Our workflow recommendations provide a way to do this in a way that is scalable and allows incremental processing of the sequencing data.
The key point is that you don’t actually have to call variants on all your samples together to perform a joint analysis. We have developed a workflow that allows us to decouple the initial identification of potential variant sites (ie variant calling) from the genotyping step, which is the only part that really needs to be done jointly. Since GATK 3.0, you can use the HaplotypeCaller to call variants individually per-sample in -ERC GVCF mode, followed by a joint genotyping step on all samples in the cohort, as described in this method article. This achieves what we call incremental joint discovery, providing you with all the benefits of classic joint calling (as described below) without the drawbacks.
Why "almost always"? Because some people have reported missing a small fraction of singletons (variants that are unique to individual samples) when using the new method. For most studies, this is an acceptable tradeoff (which is reduced by the availability of high quality sequencing data), but if you are very specifically looking for singletons, you may need to do some careful evaluation before committing to this method.
Until recently, three strategies were available for variant discovery in multiple samples:
- single sample calling: sample BAMs are analyzed individually, and individual call sets are combined in a downstream processing step;
- batch calling: sample BAMs are analyzed in separate batches, and batch call sets are merged in a downstream processing step;
- joint calling: variants are called simultaneously across all sample BAMs, generating a single call set for the entire cohort.
The best of these, from the point of view of variant discovery, was joint calling, because it provided the following benefits:
Batch-calling does not output a genotype call at sites where no member in the batch has evidence for a variant; it is thus impossible to distinguish such sites from locations missing data. In contrast, joint calling emits genotype calls at every site where any individual in the call set has evidence for variation.
By sharing information across all samples, joint calling makes it possible to “rescue” genotype calls at sites where a carrier has low coverage but other samples within the call set have a confident variant at that location. However this does not apply to singletons, which are unique to a single sample. To minimize the chance of missing singletons, we increase the cohort size -- so that singletons themselves have less chance of happening in the first place.
The current approaches to variant filtering (such as VQSR) use statistical models that work better with large amounts of data. Of the three calling strategies above, only joint calling provides enough data for accurate error modeling and ensures that filtering is applied uniformly across all samples.

Figure 1: Power of joint calling in finding mutations at low coverage sites. The variant allele is present in only two of the N samples, in both cases with such low coverage that the variant is not callable when processed separately. Joint calling allows evidence to be accumulated over all samples and renders the variant callable. (right) Importance of joint calling to square off the genotype matrix, using an example of two disease-relevant variants. Neither sample will have records in a variants-only output file, for different reasons: the first sample is homozygous reference while the second sample has no data. However, merging the results from single sample calling will incorrectly treat both of these samples identically as being non-informative.
There are two major problems with the joint calling strategy.
- Scaling & infrastructure
Joint calling scales very badly -- the calculations involved in variant calling (especially by methods like the HaplotypeCaller’s) become exponentially more computationally costly as you add samples to the cohort. If you don't have a lot of compute available, you run into limitations pretty quickly. Even here at Broad where we have fairly ridiculous amounts of compute available, we can't brute-force our way through the numbers for the larger cohort sizes that we're called on to handle.
- The N+1 problem
When you’re getting a large-ish number of samples sequenced (especially clinical samples), you typically get them in small batches over an extended period of time, and you analyze each batch as it comes in (whether it’s because the analysis is time-sensitive or your PI is breathing down your back). But that’s not joint calling, that’s batch calling, and it doesn’t give you the same significant gains that joint calling can give you. Unfortunately the joint calling approach doesn’t allow for incremental analysis -- every time you get even one new sample sequence, you have to re-call all samples from scratch.
I ran the following command
java -Xmx130g -Xms80g -Djava.io.tmpdir=javatmp/ -jar ~/picard.jar CollectHsMetrics BAIT_INTERVALS=annotations/NexteraRapidCapture_Exome_Probes_v1.2.interval_list TARGET_INTERVALS=annotations/nexterarapidcapture_exome_targetedregions_v1.2.no_chr.MT.interval_list INPUT=sample.bam OUTPUT=sample.hsmetrics METRIC_ACCUMULATION_LEVEL=ALL_READS QUIET=true VALIDATION_STRINGENCY=SILENT 2> sample.hsmetrics.log
and the program seems got stuck at a certain step. The last line in the log file is
INFO 2017-03-30 10:15:28 TheoreticalSensitivity Calculating theoretical het sensitivity
The input bam file is from gatk LeftAlignIndels. The above command worked fine on other input files generated the same way. I also tried CalculateHsMetrics and got the same problem. I tried picard version 2.3.0 and the latest 2.9.0 with the same problem. It didn't create the expected output file and program kept running (not being idle but consuming cpu time). I'm wondering if the program could enter a endless loop in some special cases. I'd tried leaving the program running for days with no result or error messages (had to kill the program at the end).
I am attempting to run Churchill, a pipeline that is designed to speed up variant calling on whole genomes and run into the error I post below. I've read several threads on this site that discuss the same problem and most suggest there is a problem with my .vcf header. I've tried for several days but can't find where that problem might be. If anyone can help I would appreciate it.
I hesitate to post the vcf header because it's huge but will if there are no objections. Below are the error details.
Running GATK3.2.2 (as per the Churchill documentation) on an SGE cluster.
.....
java.lang.NumberFormatException: For input string: "R"
at java.lang.NumberFormatException.forInputString(NumberFormatException.java:65)
at java.lang.Integer.parseInt(Integer.java:580)
at java.lang.Integer.valueOf(Integer.java:766)
at htsjdk.variant.vcf.VCFCompoundHeaderLine.(VCFCompoundHeaderLine.java:171)
at htsjdk.variant.vcf.VCFFormatHeaderLine.(VCFFormatHeaderLine.java:49)
at htsjdk.variant.vcf.AbstractVCFCodec.parseHeaderFromLines(AbstractVCFCodec.java:211)
at htsjdk.variant.vcf.VCFCodec.readActualHeader(VCFCodec.java:111)
at htsjdk.tribble.AsciiFeatureCodec.readHeader(AsciiFeatureCodec.java:88)
at htsjdk.tribble.AsciiFeatureCodec.readHeader(AsciiFeatureCodec.java:41)
at htsjdk.tribble.index.IndexFactory$FeatureIterator.readHeader(IndexFactory.java:413)
at htsjdk.tribble.index.IndexFactory$FeatureIterator.(IndexFactory.java:401)
at htsjdk.tribble.index.IndexFactory.createDynamicIndex(IndexFactory.java:312)
at org.broadinstitute.gatk.engine.refdata.tracks.RMDTrackBuilder.createIndexInMemory(RMDTrackBuilder.java:402)
at org.broadinstitute.gatk.engine.refdata.tracks.RMDTrackBuilder.attemptToLockAndLoadIndexFromDisk(RMDTrackBuilder.java:317)
at org.broadinstitute.gatk.engine.refdata.tracks.RMDTrackBuilder.loadIndex(RMDTrackBuilder.java:279)
at org.broadinstitute.gatk.engine.refdata.tracks.RMDTrackBuilder.getFeatureSource(RMDTrackBuilder.java:225)
at org.broadinstitute.gatk.engine.refdata.tracks.RMDTrackBuilder.createInstanceOfTrack(RMDTrackBuilder.java:148)
at org.broadinstitute.gatk.engine.datasources.rmd.ReferenceOrderedQueryDataPool.(ReferenceOrderedDataSource.java:208)
at org.broadinstitute.gatk.engine.datasources.rmd.ReferenceOrderedDataSource.(ReferenceOrderedDataSource.java:88)
at org.broadinstitute.gatk.engine.GenomeAnalysisEngine.getReferenceOrderedDataSources(GenomeAnalysisEngine.java:990)
at org.broadinstitute.gatk.engine.GenomeAnalysisEngine.initializeDataSources(GenomeAnalysisEngine.java:772)
at org.broadinstitute.gatk.engine.GenomeAnalysisEngine.execute(GenomeAnalysisEngine.java:285)
at org.broadinstitute.gatk.engine.CommandLineExecutable.execute(CommandLineExecutable.java:121)
at org.broadinstitute.gatk.utils.commandline.CommandLineProgram.start(CommandLineProgram.java:248)
at org.broadinstitute.gatk.utils.commandline.CommandLineProgram.start(CommandLineProgram.java:155)
at org.broadinstitute.gatk.engine.CommandLineGATK.main(CommandLineGATK.java:107)
Hi, I got an error during VariantFiltration step stating " GATK_All_Variants.vcf and reference have incompatible contigs. Please see https://software.broadinstitute.org/gatk/documentation/article?id=63for more information." i cheked the link and it suggested to remap using the right reference. However I have used the same reference for mapping that I am providing for variantFiltration. Along the way I have just used the training set for base recalibration before calling the variant. Can you suggest something?
Dear GATK team.
I am using Mutect2 to call somatic mutation from tumor/normal paired sample. However after jobs running for 8 days, our server has been rebooted for some reason. Most of the jobs are done by more than 70%. For example, some jobs called variants at Chr14, some at Chr19, and it seems the variants calling are by chromosomes. May I ask is there a way to continue the unfinished part?
I used parallel option (-nct 4) and nonparallel option for the same jobs, but it turns out that the system used more time on communication among different threads, rather than actually speeding up. Parallel jobs are actually about 4 times slower than non-parallel jobs. Considering garbage collection problem, I added java -Xmx24G -XX:+UseConcMarkSweepGC -XX:ParallelGCThreads=4 ... in the command.
Could I submit Mutect2 for each chromosome, rather than whole genome? By submitting jobs for each chromosome can actually make the variants calling parallel.
Thanks,
Qingrun
We are working on the FPGA accelerating in BWA, does GATK4 open the relative port? THX.
Hello, I am currently working on a benchmark analysis using different variant calling methods, including your HaplotypeCaller algorithm, using version 3.7. However the results I am getting from your algorithm show very few variants in comparison with the other tools I am using. I am not sure if any default parameters are limiting the sensitivity of the algorithm. Everything is being done with simulated reads from dwgsim program (which besides generating the reads in fq format, it also generates the vcf catalog with the simulated variants). The data points generated by gatk are around half if not less than the ones generated with the other variant calling tools.
I am leaving everything by default in the HaplotypeCaller except for: genotyping_mode DISCOVERY; -stand_call_conf 0; -dt NONE.
Thank you for your help
Hello,
Can someone tell me that what should i define gender for plant sample in gendermap file.
Please explain me??
Thank you in Advance
I have two questions with regard to RGQ and the --includeNonVariantSites flag in GenotypeGVCFs:
1) I have read in another thread that GQ and RGQ are capped at 99. However, I am seeing values that go higher than this in my VCF. I wanted to check to make sure this was not indicative of a problem with the VCF.
2) I have also noticed that for sites with RGQ annotations (those sites that were determined to be monomorphic), if a genotype is uncalled (has a value of "./."), then there are fewer fields in the sample genotype blocks than there are in the genotype format field (column 9). Is this intentional?
For instance, here are the first 14 columns from one line that contains both questions--the second genotype only has three fields and the third, fourth and fifth genotypes have RGQ values of 102:
ABCF3 699 . T . 20.51 . AN=436;DP=34798;InbreedingCoeff=-0.1104 GT:AD:DP:RGQ 0/0:176,0:176:0 ./.:195,0:195 0/0:35,0:35:102 0/0:40,0:40:102 0/0:34,0:34:102
Are these things something that I should worry about? I'm using GATK nightly-2017-10-17-g1994025, and the command I used to genotype 283 gVCFs was:
java -Xmx200G -jar ./GenomeAnalysisTK.jar -T GenotypeGVCFs -nt 44 --includeNonVariantSites -R reference.fasta --variant allGVCFfiles.bqsr.list -o samples.bqsr.raw.allSites.vcf > allSites.log 2>&1
Thanks very much for the great work you do!
Hi GATK team!
we have an issue with running the RealignerTargetCreator unfortunately. Commandline looks like this:
gatk -T RealignerTargetCreator -R ref.fasta -I /testsample.sorted.bam -nt 32 -o /testsample.intervals
INFO 13:00:59,111 HelpFormatter - ---------------------------------------------------------------------------------------------
INFO 13:00:59,141 HelpFormatter - The Genome Analysis Toolkit (GATK) vnightly-2017-07-11-g1f763d5, Compiled 2017/07/11 00:01:14
INFO 13:00:59,141 HelpFormatter - Copyright (c) 2010-2016 The Broad Institute
INFO 13:00:59,142 HelpFormatter - For support and documentation go to https://software.broadinstitute.org/gatk
INFO 13:00:59,142 HelpFormatter - [Thu Jul 20 13:00:58 UTC 2017] Executing on Linux 3.10.0-327.3.1.el7.x86_64 amd64
INFO 13:00:59,142 HelpFormatter - Java HotSpot(TM) 64-Bit Server VM 1.8.0_131-b11
INFO 13:00:59,170 HelpFormatter - Program Args: -T RealignerTargetCreator -R ref.fasta -I /testsample.sorted.bam -nt 32 -o /testsample.intervals
INFO 13:00:59,226 HelpFormatter - Executing as user on Linux 3.10.0-327.3.1.el7.x86_64 amd64; Java HotSpot(TM) 64-Bit Server VM 1.8.0_131-b11.
INFO 13:00:59,227 HelpFormatter - Date/Time: 2017/07/20 13:00:59
INFO 13:00:59,227 HelpFormatter - ---------------------------------------------------------------------------------------------
INFO 13:00:59,228 HelpFormatter - ---------------------------------------------------------------------------------------------
ERROR StatusLogger Unable to create class org.apache.logging.log4j.core.impl.Log4jContextFactory specified in jar:file:/opt/gatk/GenomeAnalysisTK.jar!/META-INF/log4j-provider.properties
ERROR StatusLogger Log4j2 could not find a logging implementation. Please add log4j-core to the classpath. Using SimpleLogger to log to the console…
After this, the application unfortunately hangs. Running this with GATK v3.7 stable is also not working, we had issues with the bug in HaplotypeCallers VectorHMM library. Any ideas what we can do?
I got some vcf result using mutect. but I have some question about the result.
the number of ref, alt genotype is 109, 29, but why alllele frequency is 0.078 ?
is it correct that AF 0.21 ?? (29 / (109+29))
I don't understand.
below is basecount (gatk) result of mutect bamout file.
chr12:93966398 186 93.00 58 A:38 C:20 G:0 T:0 N:0 128 A:96 C:32 G:0 T:0 N:0
vcf result indicate ref(A) =90 and alt(C)=32, but mutect bamout file indicate different basecount (A=96, C=32).
why is the number of basecount difference between vcf and mutect bam?
please answer my question.
thanks.
yh
ps. I used mutect2 and gatk3.6(DepthOfCoverage)