Document is in BETA. It may be incomplete and/or inaccurate. Post suggestions to the Comments section and be sure to read about updates also within the Comments section.
![image]() This exploratory tutorial provides instructions and example data to map short reads to a reference genome with alternate haplotypes. Instructions are suitable for indexing and mapping reads to GRCh38.
This exploratory tutorial provides instructions and example data to map short reads to a reference genome with alternate haplotypes. Instructions are suitable for indexing and mapping reads to GRCh38.
► If you are unfamiliar with terms that describe reference genome components, or GRCh38 alternate haplotypes, take a few minutes to study the Dictionary entry Reference Genome Components.
► For an introduction to GRCh38, see Blog#8180.
Specifically, the tutorial uses BWA-MEM to index and map simulated reads for three samples to a mini-reference composed of a GRCh38 chromosome and alternate contig (sections 1–3). We align in an alternate contig aware (alt-aware) manner, which we also call alt-handling. This is the main focus of the tutorial.
The decision to align to a genome with alternate haplotypes has implications for variant calling. We discuss these in section 5 using the callset generated with the optional tutorial steps outlined in section 4. Because we strategically placed a number of SNPs on the sequence used to simulate the reads, in both homologous and divergent regions, we can use the variant calls and their annotations to examine the implications of analysis approaches. To this end, the tutorial fast-forwards through pre-processing and calls variants for a trio of samples that represents the combinations of the two reference haplotypes (the PA and the ALT). This first workflow (tutorial_8017) is suitable for calling variants on the primary assembly but is insufficient for capturing variants on the alternate contigs.
For those who are interested in calling variants on the alternate contigs, we also present a second and a third workflow in section 6. The second workflow (tutorial_8017_toSE) takes the processed BAM from the first workflow, makes some adjustments to the reads to maximize their information, and calls variants on the alternate contig. This approach is suitable for calling on ~75% of the non-HLA alternate contigs or ~92% of loci with non-HLA alternate contigs (see table in section 6). The third workflow (tutorial_8017_postalt) takes the alt-aware alignments from the first workflow and performs a postalt-processing step as well as the same adjustment from the second workflow. Postalt-processing uses the bwa-postalt.js javascript program that Heng Li provides as a companion to BWA. This allows for variant calling on all alternate contigs including HLA alternate contigs.
The tutorial ends by comparing the difference in call qualities from the multiple workflows for the given example data and discusses a few caveats of each approach.
► The three workflows shown in the diagram above are available as WDL scripts in our GATK Tutorials WDL scripts repository.
Jump to a section
- Index the reference FASTA for use with BWA-MEM
- Include the reference ALT index file
☞ What happens if I forget the ALT index file?
- Align reads with BWA-MEM
☞ How can I tell if a BAM was aligned with alt-handling?
☞ What is the pa tag?
- (Optional) Add read group information, preprocess to make a clean BAM and call variants
- How can I tell whether I should consider an alternate haplotype for a given sample?
(5.1) Discussion of variant calls for tutorial_8017
- My locus includes an alternate haplotype. How can I call variants on alt contigs?
(6.1) Variant calls for tutorial_8017_toSE
(6.2) Variant calls for tutorial_8017_postalt
- Related resources
Tools involved
BWA v0.7.13 or later releases. The tutorial uses v0.7.15.
Download from here and see Tutorial#2899 for installation instructions.
The bwa-postalt.js script is within the bwakit folder.
Picard tools v2.5.0 or later releases. The tutorial uses v2.5.0.
- Optional GATK tools. The tutorial uses v3.6.
- Optional Samtools. The tutorial uses v1.3.1.
- Optional Gawk, an AWK-like tool that can interpret bitwise SAM flags. The tutorial uses v4.1.3.
- Optional k8 Javascript shell. The tutorial uses v0.2.3 downloaded from here.
Download example data
Download tutorial_8017.tar.gz, either from the GoogleDrive or from the ftp site. To access the ftp site, leave the password field blank. The data tarball contains the paired FASTQ reads files for three samples. It also contains a mini-reference chr19_chr19_KI270866v1_alt.fasta and corresponding .dict dictionary, .fai index and six BWA indices including the .alt index. The data tarball includes the output files from the workflow that we care most about. These are the aligned SAMs, processed and indexed BAMs and the final multisample VCF callsets from the three presented workflows.
![image]() The mini-reference contains two contigs subset from human GRCh38:
The mini-reference contains two contigs subset from human GRCh38: chr19 and chr19_KI270866v1_alt. The ALT contig corresponds to a diverged haplotype of chromosome 19. Specifically, it corresponds to chr19:34350807-34392977, which contains the glucose-6-phosphate isomerase or GPI gene. Part of the ALT contig introduces novel sequence that lacks a corresponding region in the primary assembly.
Using instructions in Tutorial#7859, we simulated paired 2x151 reads to derive three different sample reads that when aligned give roughly 35x coverage for the target primary locus. We derived the sequences from either the 43 kbp ALT contig (sample ALTALT), the corresponding 42 kbp region of the primary assembly (sample PAPA) or both (sample PAALT). Before simulating the reads, we introduced four SNPs to each contig sequence in a deliberate manner so that we can call variants.
► Alternatively, you may instead use the example input files and commands with the full GRCh38 reference. Results will be similar with a handful of reads mapping outside of the mini-reference regions.
1. Index the reference FASTA for use with BWA-MEM
Our example chr19_chr19_KI270866v1_alt.fasta reference already has chr19_chr19_KI270866v1_alt.dict dictionary and chr19_chr19_KI270866v1_alt.fasta.fai index files for use with Picard and GATK tools. BWA requires a different set of index files for alignment. The command below creates five of the six index files we need for alignment. The command calls the index function of BWA on the reference FASTA.
bwa index chr19_chr19_KI270866v1_alt.fasta
This gives .pac, .bwt, .ann, .amb and .sa index files that all have the same chr19_chr19_KI270866v1_alt.fasta basename. Tools recognize index files within the same directory by their identical basename. In the case of BWA, it uses the basename preceding the .fasta suffix and searches for the index file, e.g. with .bwt suffix or .64.bwt suffix. Depending on which of the two choices it finds, it looks for the same suffix for the other index files, e.g. .alt or .64.alt. Lack of a matching .alt index file will cause BWA to map reads without alt-handling. More on this next.
Note that the .64. part is an explicit indication that index files were generated with version 0.6 or later of BWA and are the 64-bit indices (as opposed to files generated by earlier versions, which were 32-bit). This .64. signifier can be added automatically by adding -6 to the bwa index command.
back to top
2. Include the reference ALT index file
Be sure to place the tutorial's mini-ALT index file chr19_chr19_KI270866v1_alt.fasta.alt with the other index files. Also, if it does not already match, change the file basename to match. This is the sixth index file we need for alignment. BWA-MEM uses this file to prioritize primary assembly alignments for reads that can map to both the primary assembly and an alternate contig. See BWA documentation for details.
As of this writing (August 8, 2016), the SAM format ALT index file for GRCh38 is available only in the x86_64-linux bwakit download as stated in this bwakit README. The hs38DH.fa.alt file is in the resource-GRCh38 folder.
In addition to mapped alternate contig records, the ALT index also contains decoy contig records as unmapped SAM records. This is relevant to the postalt-processing we discuss in section 6.2. As such, the postalt-processing in section 6 also requires the ALT index.
For the tutorial, we subset from hs38DH.fa.alt to create a mini-ALT index, chr19_chr19_KI270866v1_alt.fasta.alt. Its contents are shown below.
![image]()
The record aligns the chr19_KI270866v1_alt contig to the chr19 locus starting at position 34,350,807 and uses CIGAR string nomenclature to indicate the pairwise structure. To interpret the CIGAR string, think of the primary assembly as the reference and the ALT contig sequence as the read. For example, the 11307M at the start indicates 11,307 corresponding sequence bases, either matches or mismatches. The 935S at the end indicates a 935 base softclip for the ALT contig sequence that lacks corresponding sequence in the primary assembly. This is a region that we consider highly divergent or novel. Finally, notice the NM tag that notes the edit distance to the reference.
☞ What happens if I forget the ALT index file?
If you omit the ALT index file from the reference, or if its naming structure mismatches the other indexes, then your alignments will be equivalent to the results you would obtain if you run BWA-MEM with the -j option. The next section gives an example of what this looks like.
back to top
3. Align reads with BWA-MEM
The command below uses an alt-aware version of BWA and maps reads using BWA's maximal exact match (MEM) option. Because the ALT index file is present, the tool prioritizes mapping to the primary assembly over ALT contigs. In the command, the tutorial's chr19_chr19_KI270866v1_alt.fasta serves as reference; one FASTQ holds the forward reads and the other holds the reverse reads.
bwa mem chr19_chr19_KI270866v1_alt.fasta 8017_read1.fq 8017_read2.fq > 8017_bwamem.sam
The resulting file 8017_bwamem.sam contains aligned read records.
BWA preferentially maps to the primary assembly any reads that can align equally well to the primary assembly or the ALT contigs as well as any reads that it can reasonably align to the primary assembly even if it aligns better to an ALT contig. Preference is given by the primary alignment record status, i.e. not secondary and not supplementary. BWA takes the reads that it cannot map to the primary assembly and attempts to map them to the alternate contigs. If a read can map to an alternate contig, then it is mapped to the alternate contig as a primary alignment. For those reads that can map to both and align better to the ALT contig, the tool flags the ALT contig alignment record as supplementary (0x800). This is what we call alt-aware mapping or alt-handling.
Adding the -j option to the command disables the alt-handling. Reads that can map multiply are given low or zero MAPQ scores.
![image]()
☞ How can I tell if a BAM was aligned with alt-handling?
There are two approaches to this question.
First, you can view the alignments on IGV and compare primary assembly loci with their alternate contigs. The IGV screenshots to the right show how BWA maps reads with (top) or without (bottom) alt-handling.
Second, you can check the alignment SAM. Of two tags that indicate alt-aware alignment, one will persist after preprocessing only if the sample has reads that can map to alternate contigs. The first tag, the AH tag, is in the BAM header section of the alignment file, and is absent after any merging step, e.g. merging with MergeBamAlignment. The second tag, the pa tag, is present for reads that the aligner alt-handles. If a sample does not contain any reads that map equally or preferentially to alternate contigs, then this tag may be absent in a BAM even if the alignments were mapped in an alt-aware manner.
Here are three headers for comparison where only one indicates alt-aware alignment.
File header for alt-aware alignment. We use this type of alignment in the tutorial.
Each alternate contig's @SQ line in the header will have an AH:* tag to indicate alternate contig handling for that contig. This marking is based on the alternate contig being listed in the .alt index file and alt-aware alignment.
![image]()
File header for -j alignment (alt-handling disabled) for example purposes. We do not perform this type of alignment in the tutorial.
Notice the absence of any special tags in the header.
![image]()
File header for alt-aware alignment after merging with MergeBamAlignment. We use this step in the next section.
Again, notice the absence of any special tags in the header.
![image]()
☞ What is the pa tag?
For BWA v0.7.15, but not v0.7.13, ALT loci alignment records that can align to both the primary assembly and alternate contig(s) will have a pa tag on the primary assembly alignment. For example, read chr19_KI270866v1_alt_4hetvars_26518_27047_0:0:0_0:0:0_931 of the ALTALT sample has five alignment records only three of which have the pa tag as shown below.
![image]()
A brief description of each of the five alignments, in order:
- First in pair, primary alignment on the primary assembly; AS=146, pa=0.967
- First in pair, supplementary alignment on the alternate contig; AS=151
- Second in pair, primary alignment on the primary assembly; AS=120; pa=0.795
- Second in pair, supplementary alignment on the primary assembly; AS=54; pa=0.358
- Second in pair, supplementary alignment on the alternate contig; AS=151
The pa tag measures how much better a read aligns to its best alternate contig alignment versus its primary assembly (pa) alignment. Specifically, it is the ratio of the primary assembly alignment score over the highest alternate contig alignment score. In our example we have primary assembly alignment scores of 146, 120 and 54 and alternate contig alignment scores of 151 and again 151. This gives us three different pa scores that tag the primary assembly alignments: 146/151=0.967, 120/151=0.795 and 54/151=0.358.
In our tutorial's workflow, MergeBamAlignment may either change an alignment's pa score or add a previously unassigned pa score to an alignment. The result of this is summarized as follows for the same alignments.
- pa=0.967 --MergeBamAlignment--> same
- none --MergeBamAlignment--> assigns pa=0.967
- pa=0.795 --MergeBamAlignment--> same
- pa=0.358 --MergeBamAlignment--> replaces with pa=0.795
- none --MergeBamAlignment--> assigns pa=0.795
If you want to retain the BWA-assigned pa scores, then add the following options to the workflow commands in section 4.
For RevertSam, add ATTRIBUTE_TO_CLEAR=pa.
For MergeBamAlignment, add ATTRIBUTES_TO_RETAIN=pa.
In our sample set, after BWA-MEM alignment ALTALT has 1412 pa-tagged alignment records, PAALT has 805 pa-tagged alignment records and PAPA has zero pa-tagged records.
back to top
4. Add read group information, preprocess to make a clean BAM and call variants
The initial alignment file is missing read group information. One way to add that information, which we use in production, is to use MergeBamAlignment. MergeBamAlignment adds back read group information contained in an unaligned BAM and adjusts meta information to produce a clean BAM ready for pre-processing (see Tutorial#6483 for details on our use of MergeBamAlignment). Given the focus here is to showcase BWA-MEM's alt-handling, we refrain from going into the details of all this additional processing. They follow, with some variation, the PairedEndSingleSampleWf pipeline detailed here.
Remember these are simulated reads with simulated base qualities. We simulated the reads in a manner that only introduces the planned mismatches, without any errors. Coverage is good at roughly 35x. All of the base qualities for all of the reads are at I, which is, according to this page and this site, an excellent base quality score equivalent to a Sanger Phred+33 score of 40. We can therefore skip base quality score recalibration (BQSR) since the reads are simulated and the dataset is not large enough for recalibration anyway.
Here are the commands to obtain a final multisample variant callset. The commands are given for one of the samples. Process each of the three samples independently in the same manner [4.1–4.6] until the last GenotypeGVCFs command [4.7].
[4.1] Create unmapped uBAM
java -jar picard.jar RevertSam \
I=altalt_bwamem.sam O=altalt_u.bam \
ATTRIBUTE_TO_CLEAR=XS ATTRIBUTE_TO_CLEAR=XA
[4.2] Add read group information to uBAM
java -jar picard.jar AddOrReplaceReadGroups \
I=altalt_u.bam O=altalt_rg.bam \
RGID=altalt RGSM=altalt RGLB=wgsim RGPU=shlee RGPL=illumina
[4.3] Merge uBAM with aligned BAM
java -jar picard.jar MergeBamAlignment \
ALIGNED=altalt_bwamem.sam UNMAPPED=altalt_rg.bam O=altalt_m.bam \
R=chr19_chr19_KI270866v1_alt.fasta \
SORT_ORDER=unsorted CLIP_ADAPTERS=false \
ADD_MATE_CIGAR=true MAX_INSERTIONS_OR_DELETIONS=-1 \
PRIMARY_ALIGNMENT_STRATEGY=MostDistant \
UNMAP_CONTAMINANT_READS=false \
ATTRIBUTES_TO_RETAIN=XS ATTRIBUTES_TO_RETAIN=XA
[4.4] Flag duplicate reads
java -jar picard.jar MarkDuplicates \
INPUT=altalt_m.bam OUTPUT=altalt_md.bam METRICS_FILE=altalt_md.bam.txt \
OPTICAL_DUPLICATE_PIXEL_DISTANCE=2500 ASSUME_SORT_ORDER=queryname
[4.5] Coordinate sort, fix NM and UQ tags and index for clean BAM
As of Picard v2.7.0, released October 17, 2016, SetNmAndUqTags is no longer available. Use SetNmMdAndUqTags instead.
set -o pipefail
java -jar picard.jar SortSam \
INPUT=altalt_md.bam OUTPUT=/dev/stdout SORT_ORDER=coordinate | \
java -jar $PICARD SetNmAndUqTags \
INPUT=/dev/stdin OUTPUT=altalt_snaut.bam \
CREATE_INDEX=true R=chr19_chr19_KI270866v1_alt.fasta
[4.6] Call SNP and indel variants in emit reference confidence (ERC) mode per sample using HaplotypeCaller
java -jar GenomeAnalysisTK.jar -T HaplotypeCaller \
-R chr19_chr19_KI270866v1_alt.fasta \
-o altalt.g.vcf -I altalt_snaut.bam \
-ERC GVCF --max_alternate_alleles 3 --read_filter OverclippedRead \
--emitDroppedReads -bamout altalt_hc.bam
[4.7] Call genotypes on three samples
java -jar GenomeAnalysisTK.jar -T GenotypeGVCFs \
-R chr19_chr19_KI270866v1_alt.fasta -o multisample.vcf \
--variant altalt.g.vcf --variant altpa.g.vcf --variant papa.g.vcf
The altalt_snaut.bam, HaplotypeCaller's altalt_hc.bam and the multisample multisample.vcf are ready for viewing on IGV.
Before getting into the results in the next section, we have minor comments on two filtering options.
In our tutorial workflows, we turn off MergeBamAlignment's UNMAP_CONTAMINANT_READS option. If set to true, 68 reads become unmapped for PAPA and 40 reads become unmapped for PAALT. These unmapped reads are those reads caught by the UNMAP_CONTAMINANT_READS filter and their mates. MergeBamAlignment defines contaminant reads as those alignments that are overclipped, i.e. that are softclipped on both ends, and that align with less than 32 bases. Changing the MIN_UNCLIPPED_BASES option from the default of 32 to 22 and 23 restores all of these reads for PAPA and PAALT, respectively. Contaminants are obviously absent for these simulated reads. And so we set UNMAP_CONTAMINANT_READS to false to disable this filtering.
HaplotypeCaller's --read_filter OverclippedRead option similarly looks for both-end-softclipped alignments, then filters reads aligning with less than 30 bases. The difference is that HaplotypeCaller only excludes the overclipped alignments from its calling and does not remove mapping information nor does it act on the mate of the filtered alignment. Thus, we keep this read filter for the first workflow. However, for the second and third workflows in section 6, tutorial_8017_toSE and tutorial_8017_postalt, we omit the --read_filter Overclipped option from the HaplotypeCaller command. We also omit the --max_alternate_alleles 3 option for simplicity.
back to top
5. How can I tell whether I should consider an alternate haplotype?
![image]() We consider this question only for our GPI locus, a locus we know has an alternate contig in the reference. Here we use the term locus in its biological sense to refer to a contiguous genomic region of interest. The three samples give the alignment and coverage profiles shown on the right.
We consider this question only for our GPI locus, a locus we know has an alternate contig in the reference. Here we use the term locus in its biological sense to refer to a contiguous genomic region of interest. The three samples give the alignment and coverage profiles shown on the right.
What is immediately apparent from the IGV screenshot is that the scenarios that include the alternate haplotype give a distinct pattern of variant sites to the primary assembly much like a fingerprint. These variants are predominantly heterozygous or homozygous. Looking closely at the 3' region of the locus, we see some alignment coverage anomalies that also show a distinct pattern. The coverage in some of the highly diverged region in the primary assembly drops while in others it increases. If we look at the origin of simulated reads in one of the excess coverage regions, we see that they are from two different regions of the alternate contig that suggests duplicated sequence segments within the alternate locus.
The variation pattern and coverage anomalies on the primary locus suggest an alternate haplotype may be present for the locus. We can then confirm the presence of aligned reads, both supplementary and primary, on the alternate locus. Furthermore, if we count the alignment records for each region, e.g. using samtools idxstats, we see the following metrics.
ALT/ALT PA/ALT PA/PA
chr19 10005 10006 10000
chr19_KI270866v1_alt 1407 799 0
The number of alignments on the alternate locus increases proportionately with alternate contig dosage. All of these factors together suggest that the sample presents an alternate haplotype.
5.1 Discussion of variant calls for tutorial_8017
The three-sample variant callset gives 54 sites on the primary locus and two additional on the alternate locus for 56 variant sites. All of the eight SNP alleles we introduced are called, with six called on the primary assembly and two called on the alternate contig. Of the 15 expected genotype calls, four are incorrect. Namely, four PAALT calls that ought to be heterozygous are called homozygous variant. These are two each on the primary assembly and on the alternate contig in the region that is highly divergent.
► Our production pipelines use genomic intervals lists that exclude GRCh38 alternate contigs from variant calling. That is, variant calling is performed only for contigs of the primary assembly. This calling on even just the primary assembly of GRCh38 brings improvements to analysis results over previous assemblies. For example, if we align and call variants for our simulated reads on GRCh37, we call 50 variant sites with identical QUAL scores to the equivalent calls in our GRCh38 callset. However, this GRCh37 callset is missing six variant calls compared to the GRCh38 callset for the 42 kb locus: the two variant sites on the alternate contig and four variant sites on the primary assembly.
Consider the example variants on the primary locus. The variant calls from the primary assembly include 32 variant sites that are strictly homozygous variant in ALTALT and heterozygous variant in PAALT. The callset represents only those reads from the ALT that can be mapped to the primary assembly.
In contrast, the two variants in regions whose reads can only map to the alternate contig are absent from the primary assembly callset. For this simulated dataset, the primary alignments present on the alternate contig provide enough supporting reads that allow HaplotypeCaller to call the two variants. However, these variant calls have lower-quality annotation metrics than for those simulated in an equal manner on the primary assembly. We will get into why this is in section 6.
Additionally, for our PAALT sample that is heterozygous for an alternate haplotype, the genotype calls in the highly divergent regions are inaccurate. These are called homozygous variant on the primary assembly and on the alternate contig when in fact they are heterozygous variant. These calls have lower genotype scores GQ as well as lower allele depth AD and coverage DP. The table below shows the variant calls for the introduced SNP sites. In blue are the genotype calls that should be heterozygous variant but are instead called homozygous variant.
![image]()
Here is a command to select out the intentional variant sites that uses SelectVariants:
java -jar GenomeAnalysisTK.jar -T SelectVariants \
-R chr19_chr19_KI270866v1_alt.fasta \
-V multisample.vcf -o multisample_selectvariants.vcf \
-L chr19:34,383,500 -L chr19:34,389,485 -L chr19:34,391,800 -L chr19:34,392,600 \
-L chr19_KI270866v1_alt:32,700 -L chr19_KI270866v1_alt:38,700 \
-L chr19_KI270866v1_alt:41,700 -L chr19_KI270866v1_alt:42,700 \
-L chr19:34,383,486 -L chr19_KI270866v1_alt:32,714
back to top
6. My locus includes an alternate haplotype. How can I call variants on alt contigs?
If you want to call variants on alternate contigs, consider additional data processing that overcome the following problems.
Loss of alignments from filtering of overclipped reads.
HaplotypeCaller's filtering of alignments whose mates map to another contig. Alt-handling produces many of these types of reads on the alternate contigs.
- Zero MAPQ scores for alignments that map to two or more alternate contigs. HaplotypeCaller excludes these types of reads from contributing to evidence for variation.
Let us talk about these in more detail.
Ideally, if we are interested in alternate haplotypes, then we would have ensured we were using the most up-to-date analysis reference genome sequence with the latest patch fixes. Also, whatever approach we take to align and preprocess alignments, if we filter any reads as putative contaminants, e.g. with MergeBamAlignment's option to unmap cross-species contamination, then at this point we would want to fish back into the unmapped reads pool and pull out those reads. Specifically, these would have an SA tag indicating mapping to the alternate contig of interest and an FT tag indicating the reason for unmapping was because MergeBamAlignment's UNMAP_CONTAMINANT_READS option identified them as cross-species contamination. Similarly, we want to make sure not to include HaplotypeCaller's --read_filter OverclippedRead option that we use in the first workflow.
![image]() As section 5.1 shows, variant calls on the alternate contig are of low quality--they have roughly an order of magnitude lower QUAL scores than what should be equivalent variant calls on the primary assembly.
As section 5.1 shows, variant calls on the alternate contig are of low quality--they have roughly an order of magnitude lower QUAL scores than what should be equivalent variant calls on the primary assembly.
For this exploratory tutorial, we are interested in calling the introduced SNPs with equivalent annotation metrics. Whether they are called on the primary assembly or the alternate contig and whether they are called homozygous variant or heterozygous--let's say these are less important, especially given pinning certain variants from highly homologous regions to one of the loci is nigh impossible with our short reads. To this end, we will use the second workflow shown in the workflows diagram. However, because this solution is limited, we present a third workflow as well.
► We present these workflows solely for exploratory purposes. They do not represent any production workflows.
Tutorial_8017_toSE uses the processed BAM from our first workflow and allows for calling on singular alternate contigs. That is, the workflow is suitable for calling on alternate contigs of loci with only a single alternate contig like our GPI locus. Tutorial_8017_postalt uses the aligned SAM from the first workflow before processing, and requires separate processing before calling. This third workflow allows for calling on all alternate contigs, even on HLA loci that have numerous contigs per primary locus. However, the callset will not be parsimonious. That is, each alternate contig will greedily represent alignments and it is possible the same variant is called for all the alternate loci for a given primary locus as well as on the primary locus. It is up to the analyst to figure out what to do with the resulting calls.
![image]() The reason for the divide in these two workflows is in the way BWA assigns mapping quality scores (MAPQ) to multimapping reads. Postalt-processing becomes necessary for loci with two or more alternate contigs because the shared alignments between the primary locus and alternate loci will have zero MAPQ scores. Postalt-processing gives non-zero MAPQ scores to the alignment records. The table presents the frequencies of GRCh38 non-HLA alternate contigs per primary locus. It appears that ~75% of non-HLA alternate contigs are singular to ~92% of primary loci with non-HLA alternate contigs. In terms of bases on the primary assembly, of the ~75 megabases that have alternate contigs, ~64 megabases (85%) have singular non-HLA alternate contigs and ~11 megabases (15%) have multiple non-HLA alternate contigs per locus. Our tutorial's example locus falls under this majority.
The reason for the divide in these two workflows is in the way BWA assigns mapping quality scores (MAPQ) to multimapping reads. Postalt-processing becomes necessary for loci with two or more alternate contigs because the shared alignments between the primary locus and alternate loci will have zero MAPQ scores. Postalt-processing gives non-zero MAPQ scores to the alignment records. The table presents the frequencies of GRCh38 non-HLA alternate contigs per primary locus. It appears that ~75% of non-HLA alternate contigs are singular to ~92% of primary loci with non-HLA alternate contigs. In terms of bases on the primary assembly, of the ~75 megabases that have alternate contigs, ~64 megabases (85%) have singular non-HLA alternate contigs and ~11 megabases (15%) have multiple non-HLA alternate contigs per locus. Our tutorial's example locus falls under this majority.
In both alt-aware mapping and postalt-processing, alternate contig alignments have a predominance of mates that map back to the primary assembly. HaplotypeCaller, for good reason, filters reads whose mates map to a different contig. However, we know that GRCh38 artificially represents alternate haplotypes as separate contigs and BWA-MEM intentionally maps these mates back to the primary locus. For comparable calls on alternate contigs, we need to include these alignments in calling. To this end, we have devised a temporary workaround.
6.1 Variant calls for tutorial_8017_toSE
Here we are only aiming for equivalent calls with similar annotation values for the two variants that are called on the alternate contig. For the solution that we will outline, here are the results.
![image]()
Including the mate-mapped-to-other-contig alignments bolsters the variant call qualities for the two SNPs HaplotypeCaller calls on the alternate locus. We see the AD allele depths much improved for ALTALT and PAALT. Corresponding to the increase in reads, the GQ genotype quality and the QUAL score (highlighted in red) indicate higher qualities. For example, the QUAL scores increase from 332 and 289 to 2166 and 1764, respectively. We also see that one of the genotype calls changes. For sample ALTALT, we see a previous no call is now a homozygous reference call (highlighted in blue). This hom-ref call is further from the truth than not having a call as the ALTALT sample should not have coverage for this region in the primary assembly.
For our example data, tutorial_8017's callset subset for the primary assembly and tutorial_8017_toSE's callset subset for the alternate contigs together appear to make for a better callset.
What solution did we apply? As the workflow's name toSE implies, this approach converts paired reads to single end reads. Specifically, this approach takes the processed and coordinate-sorted BAM from the first workflow and removes the 0x1 paired flag from the alignments. Removing the 0x1 flag from the reads allows HaplotypeCaller to consider alignments whose mates map to a different contig. We accomplish this using a modified script of that presented in Biostars post https://www.biostars.org/p/106668/, indexing with Samtools and then calling with HaplotypeCaller as follows. Note this workaround creates an invalid BAM according to ValidateSamFile. Also, another caveat is that because HaplotypeCaller uses softclipped sequences, any overlapping regions of read pairs will count twice towards variation instead of once. Thus, this step may lead to overconfident calls in such regions.
Remove the 0x1 bitwise flag from alignments
samtools view -h altalt_snaut.bam | gawk '{printf "%s\t", $1; if(and($2,0x1))
{t=$2-0x1}else{t=$2}; printf "%s\t" , t; for (i=3; i<NF; i++){printf "%s\t", $i} ;
printf "%s\n",$NF}'| samtools view -Sb - > altalt_se.bam
Index the resulting BAM
samtools index altalt_se.bam
Call variants in -ERC GVCF mode with HaplotypeCaller for each sample
java -jar GenomeAnalysisTK.jar -T HaplotypeCaller \
-R chr19_chr19_KI270866v1_alt.fasta \
-I altalt_se.bam -o altalt_hc.g.vcf \
-ERC GVCF --emitDroppedReads -bamout altalt_hc.bam
Finally, use GenotypeGVCFs as shown in section 4's command [4.7] for a multisample variant callset. Tutorial_8017_toSE calls 68 variant sites--66 on the primary assembly and two on the alternate contig.
6.2 Variant calls for tutorial_8017_postalt
BWA's postalt-processing requires the query-grouped output of BWA-MEM. Piping an alignment step with postalt-processing is possible. However, to be able to compare variant calls from an identical alignment, we present the postalt-processing as an add-on workflow that takes the alignment from the first workflow.
The command uses the bwa-postalt.js script, which we run through k8, a Javascript execution shell. It then lists the ALT index, the aligned SAM altalt.sam and names the resulting file > altalt_postalt.sam.
k8 bwa-postalt.js \
chr19_chr19_KI270866v1_alt.fasta.alt \
altalt.sam > altalt_postalt.sam
![image]() The resulting postalt-processed SAM,
The resulting postalt-processed SAM, altalt_postalt.sam, undergoes the same processing as the first workflow (commands 4.1 through 4.7) except that (i) we omit --max_alternate_alleles 3 and --read_filter OverclippedRead options for the HaplotypeCaller command like we did in section 6.1 and (ii) we perform the 0x1 flag removal step from section 6.1.
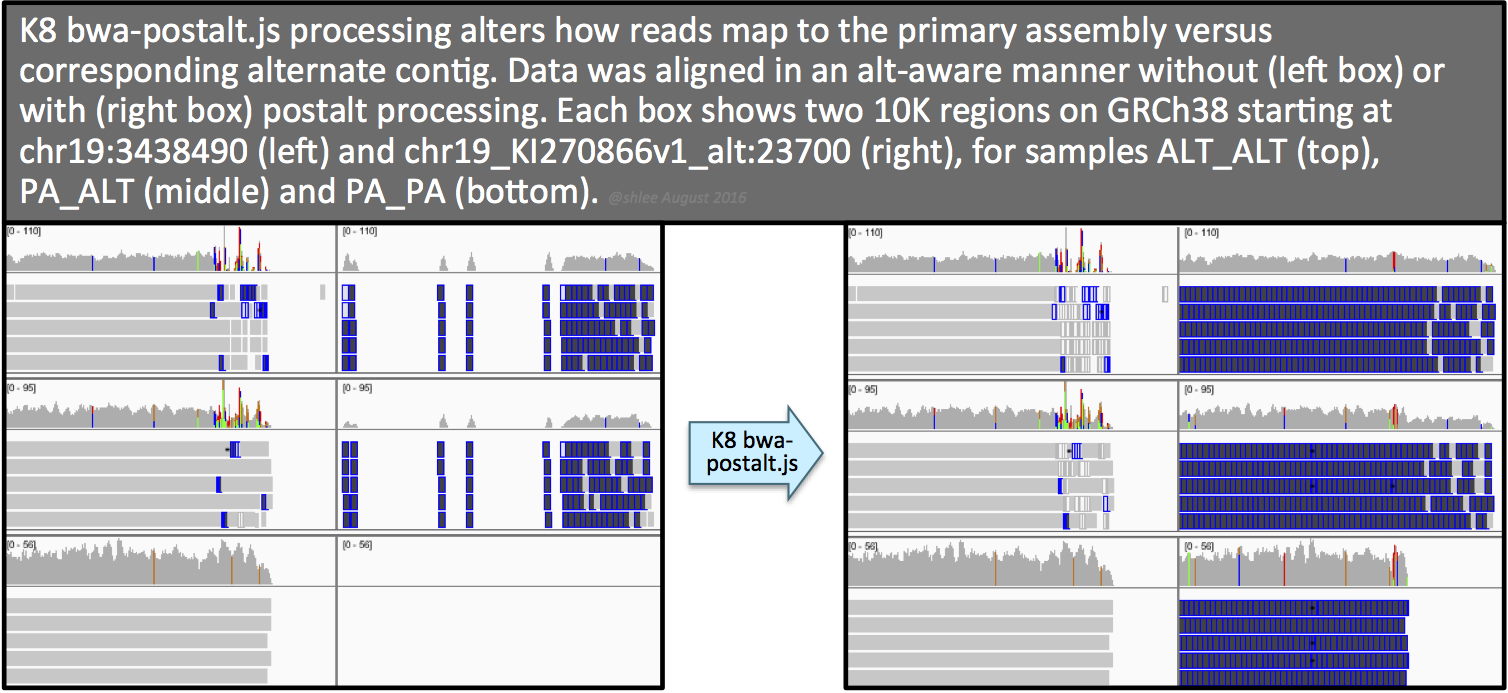
The effect of this postalt-processing is immediately apparent in the IGV screenshots. Previously empty regions are now filled with alignments. Look closely in the highly divergent region of the primary locus. Do you notice a change, albeit subtle, before and after postalt-processing for samples ALTALT and PAALT?
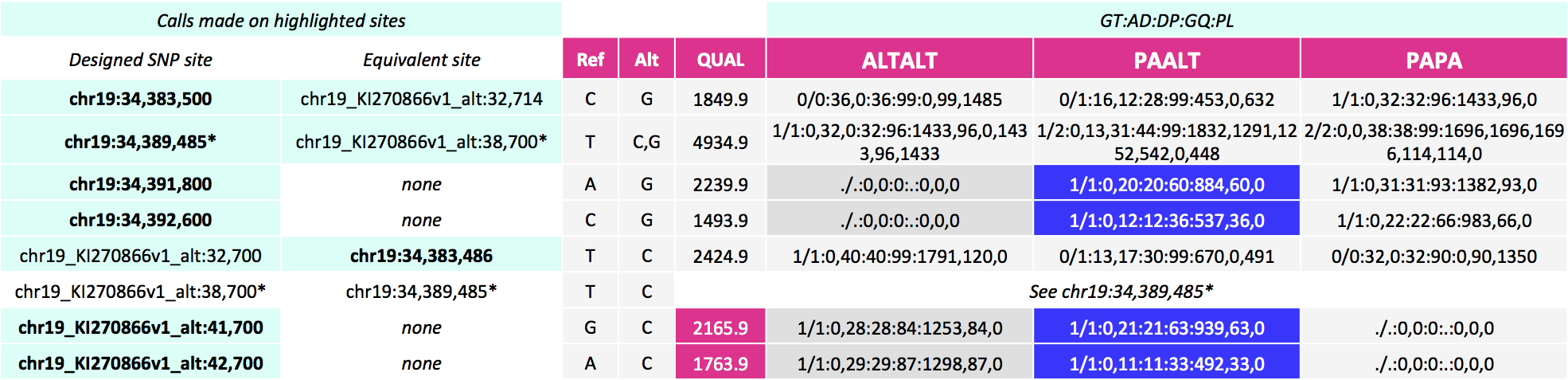
These alignments give the calls below for our SNP sites of interest. Here, notice calls are made for more sites--on the equivalent site if present in addition to the design site (highlighted in the first two columns). For the three pairs of sites that can be called on either the primary locus or alternate contig, the variant site QUALs, the INFO field annotation metrics and the sample level annotation values are identical for each pair.
![image]()
Postalt-processing lowers the MAPQ of primary locus alignments in the highly divergent region that map better to the alt locus. You can see this as a subtle change in the IGV screenshot. After postalt-processing we see an increase in white zero MAPQ reads in the highly divergent region of the primary locus for ALTALT and PAALT. For ALTALT, this effectively cleans up the variant calls in this region at chr19:34,391,800 and chr19:34,392,600. Previously for ALTALT, these calls contained some reads: 4 and 25 for the first workflow and 0 and 28 for the second workflow. After postalt-processing, no reads are considered in this region giving us ./.:0,0:0:.:0,0,0 calls for both sites.
What we omit from examination are the effects of postalt-processing on decoy contig alignments. Namely, if an alignment on the primary assembly aligns better on a decoy contig, then postalt-processing discounts the alignment on the primary assembly by assigning it a zero MAPQ score.
To wrap up, here are the number of variant sites called for the three workflows. As you can see, this last workflow calls the most variants at 95 variant sites, with 62 on the primary assembly and 33 on the alternate contig.
Workflow total on primary assembly on alternate contig
tutorial_8017 56 54 2
tutorial_8017_toSE 68 66 2
tutorial_8017_postalt 95 62 33
back to top
7. Related resources
For WDL scripts of the workflows represented in this tutorial, see the GATK WDL scripts repository.
To revert an aligned BAM to unaligned BAM, see Section B of Tutorial#6484.
- To simulate reads from a reference contig, see Tutorial#7859.
- Dictionary entry Reference Genome Components reviews terminology that describe reference genome components.
- The GATK resource bundle provides an analysis set GRCh38 reference FASTA as well as several other related resource files.
- As of this writing (August 8, 2016), the SAM format ALT index file for GRCh38 is available only in the x86_64-linux bwakit download as stated in this bwakit README. The
hs38DH.fa.alt file is in the resource-GRCh38 folder. Rename this file's basename to match that of the corresponding reference FASTA.
- For more details on MergeBamAlignment features, see Section 3C of Tutorial#6483.
- For details on the PairedEndSingleSampleWorkflow that uses GRCh38, see here.
- See here for VCF specifications.
back to top
 If you are interested in emulating the methods used by the Broad Genomics Platform to pre-process your short read sequencing data, you have landed on the right page. The parsimonious operating procedures outlined in this three-step workflow both maximize data quality, storage and processing efficiency to produce a mapped and clean BAM. This clean BAM is ready for analysis workflows that start with MarkDuplicates.
If you are interested in emulating the methods used by the Broad Genomics Platform to pre-process your short read sequencing data, you have landed on the right page. The parsimonious operating procedures outlined in this three-step workflow both maximize data quality, storage and processing efficiency to produce a mapped and clean BAM. This clean BAM is ready for analysis workflows that start with MarkDuplicates.









 A read with multiple alignment records may map to multiple loci or may be chimeric--that is, splits the alignment. It is possible for an aligner to produce multiple alignments as well as multiple primary alignments, e.g. in the case of a linear alignment set of split reads. When one alignment, or alignment set in the case of chimeric read records, is designated primary, others are designated either secondary or supplementary. Invoking the
A read with multiple alignment records may map to multiple loci or may be chimeric--that is, splits the alignment. It is possible for an aligner to produce multiple alignments as well as multiple primary alignments, e.g. in the case of a linear alignment set of split reads. When one alignment, or alignment set in the case of chimeric read records, is designated primary, others are designated either secondary or supplementary. Invoking the  Tags are updated for our example data as shown in the table. The tool retains SA, MD, NM and AS tags from the alignment, given these are not present in the uBAM. The tool additionally adds UQ (the Phred likelihood of the segment), MC (mate CIGAR string) and MQ (mapping quality of the mate/next segment) tags if applicable. For unmapped reads (marked with an
Tags are updated for our example data as shown in the table. The tool retains SA, MD, NM and AS tags from the alignment, given these are not present in the uBAM. The tool additionally adds UQ (the Phred likelihood of the segment), MC (mate CIGAR string) and MQ (mapping quality of the mate/next segment) tags if applicable. For unmapped reads (marked with an 

 We pipe the three tools described above to generate an aligned BAM file sorted by query name. In the piped command, the commands for the three processes are given together, separated by a
We pipe the three tools described above to generate an aligned BAM file sorted by query name. In the piped command, the commands for the three processes are given together, separated by a  Here we outline how to generate an unmapped BAM (uBAM) from either a FASTQ or aligned BAM file. We use Picard's FastqToSam to convert a FASTQ (Option A) or Picard's RevertSam to convert an aligned BAM (Option B).
Here we outline how to generate an unmapped BAM (uBAM) from either a FASTQ or aligned BAM file. We use Picard's FastqToSam to convert a FASTQ (Option A) or Picard's RevertSam to convert an aligned BAM (Option B).